Week 4:
Carver College of Medicine
June 16, 2014
This week I started with some minipreps with twelve different transfected cell cultures. I did minipreps to first figure out which cell transfection could be digested by the enzyme and we would proceed by making more of this type of plasmid with midipreps. It was a plasmid preparation method used to extract and purify plasmid DNA and involved three steps: growth of the E. coli culture, harvest and lysis of the cells, and purification of plasmid DNA. Minipreps were analogous to the midipreps that I did before, but they were much simpler because of their smaller scales. After minipreps were completed, I digested samples 1 – 4 with restriction enzymes AhdI and EcoRI-HF. This master mix contained 2 µL of each of the two enzymes, 6 µL of CutSmart 10X buffer, and 38 µL of water. I digested samples 5 – 12 with restriction enzymes EcoRI-HF and MfeI-HF. This master mix contained 3µL of each of the enzymes, 9 µL of CutSmart buffer, and 57 µL of water. The digestions were done by combining 2 µL of the purified plasmid DNA with 3 µL of the master mix. I also did more minipreps later this week on six new transfections. But this time I made up more of the A1 resuspension buffer by quantitatively transfer the RNase solid to the A1 buffer prior to use. RNase was very stable in solid form and could be stored at room temperature. After it was dissolved in liquid, however, it became less stable and should be stored in the cooler.
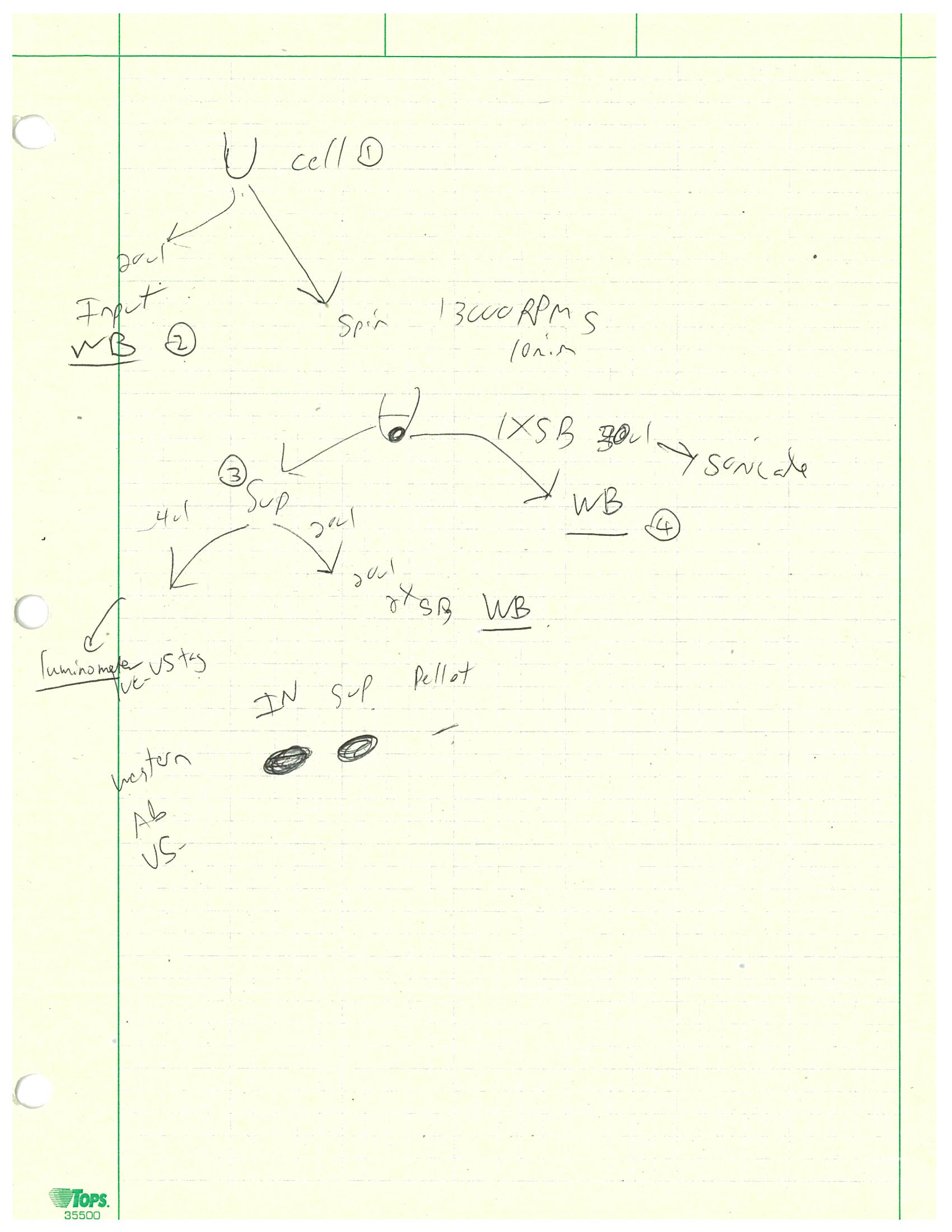
I also did more luciferase assays this week but a much more involved version. I was working on a 24-well plate, half contained PC6-3/TR-156+DOX cells and the other half HeLa cells. For each type of cell we had the wild type neurons, those with R6A mutation, and those with both K2A and R6A mutations. After washing the cells, I put 100 µL of firefly luciferase lysis buffer in each PC6-3 cell well and 70 µL in each HeLa cell well. The 4X sample buffer used in this assay contained 40 mL of Tris, 40 mL of glycerol, 8 g of SDS, and 0.1 g of bromophenol blue. 1 : 4 ratio of 2-mercaptoethanol was also added prior to use of the buffer. After making some 2X and 1X sample buffers from the 4X stock, I used large orifice tips to scrap off the cells and transfer all solutions into one set of microcentrifuge tubes. Then I took 20 µL of each sample to combine with 20 µL of 2X sample buffer into a new set of tubes. This set was the input and would be run on Western blot next week. The rest of the samples were centrifuged at 13,000 rpm for 10 minutes and all supernatants were transferred into a new set of tubes. Then 20 µL of supernatant was combined with 20 µL of 2X sample buffer into a new set of tubes which would be run on Western blot. Then 2 µL of supernatant was combined with 50 µL of luciferase substrate (with buffer) and its light intensity was measured by luminometer. Finally, 20 µL of 1X sample buffer was added to each pellet and would be run on Western blot. This entire process was outlined in the photo below.
The major project of this week, however, was the nucleus morphology assay using the Epi-fluorescence microscope. I used microscopy to inspect the number of living and dead transfected neurons in eight wells, each of which contained a different transfection treated with OGD (oxygen-glucose depravation). The eight transfections and their expected cell survival conditions were described in the table below:
Transfection Expected survival condition
Mas70-GFP (control) No change
Mff-A (0000) More death
Mff-A-VPE2X Less death
Mff-D (0011) (control) No change
Mff-D-9A Less death
Mff-D-9ED More death
Mff-D-4E More death, but not as dramatic
Mff-D-5ED More death, but not as dramatic
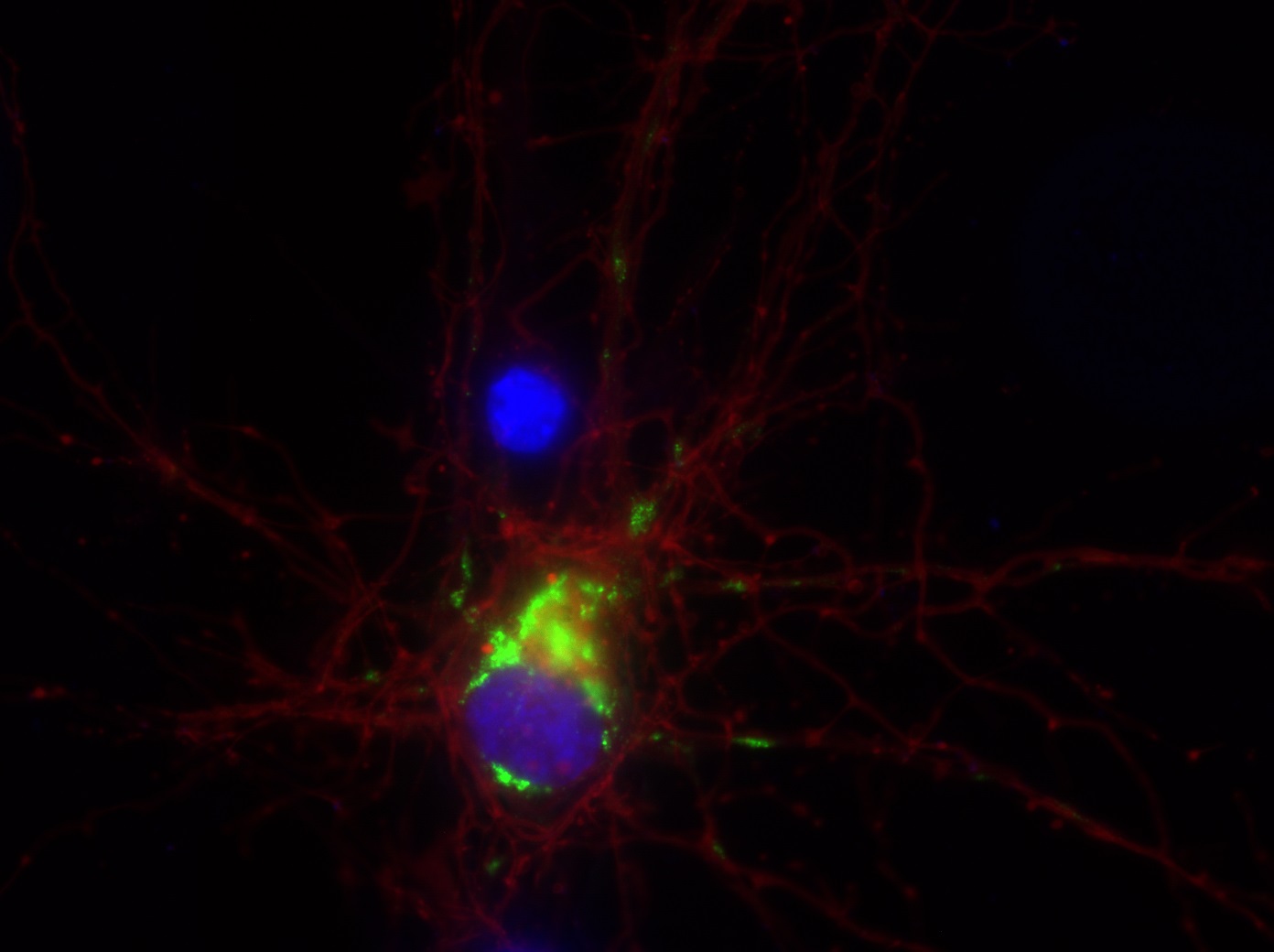
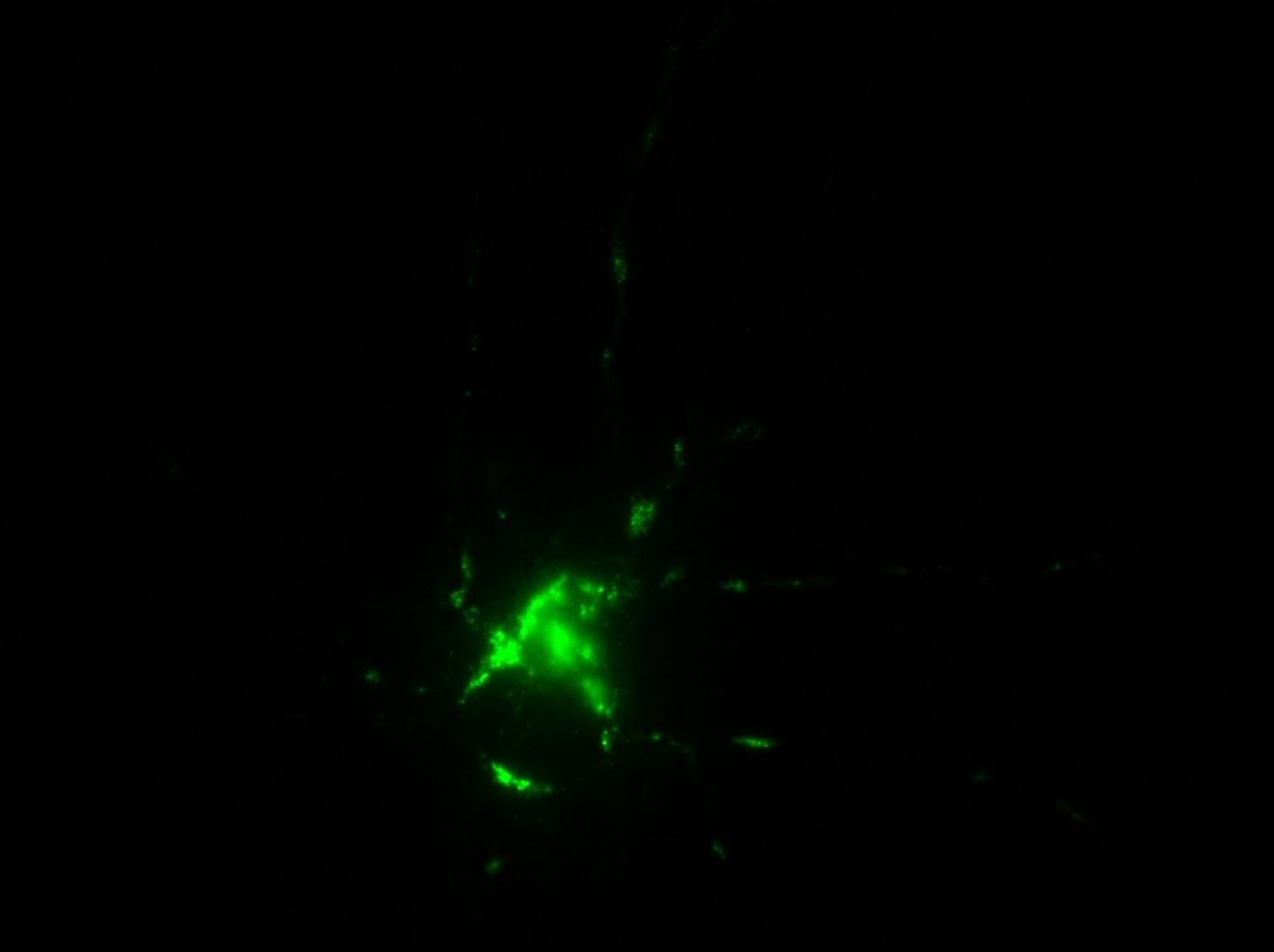
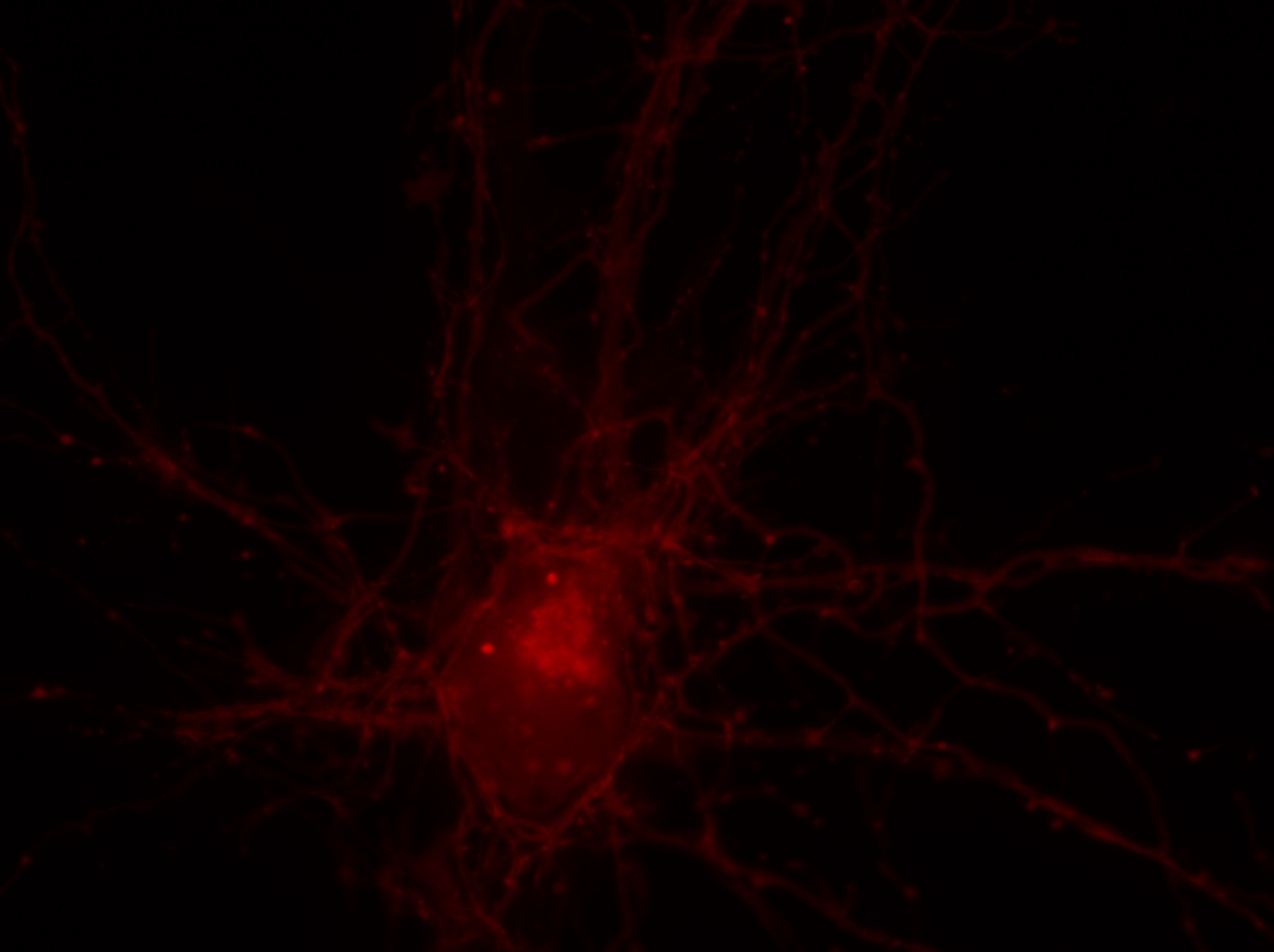
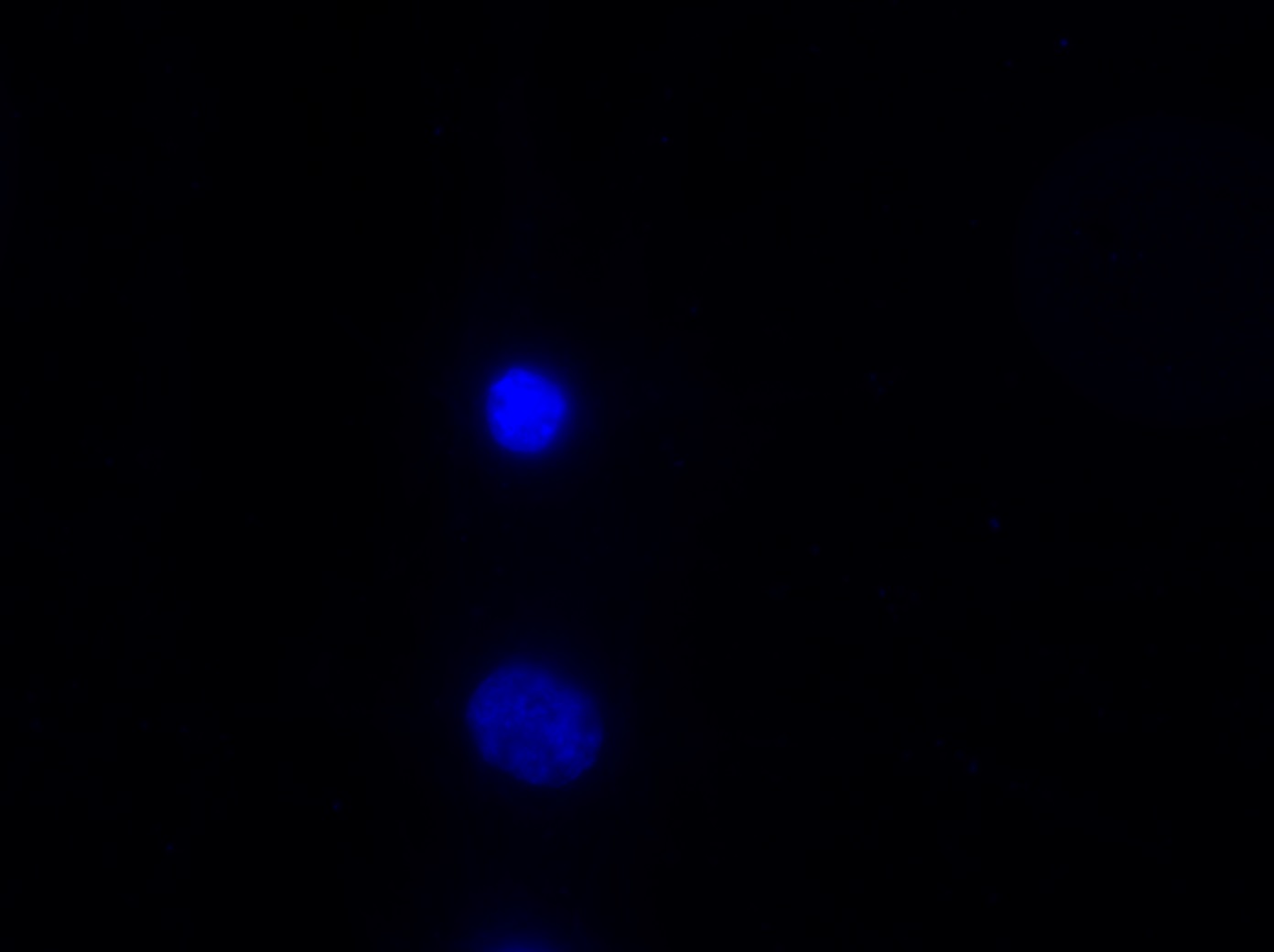
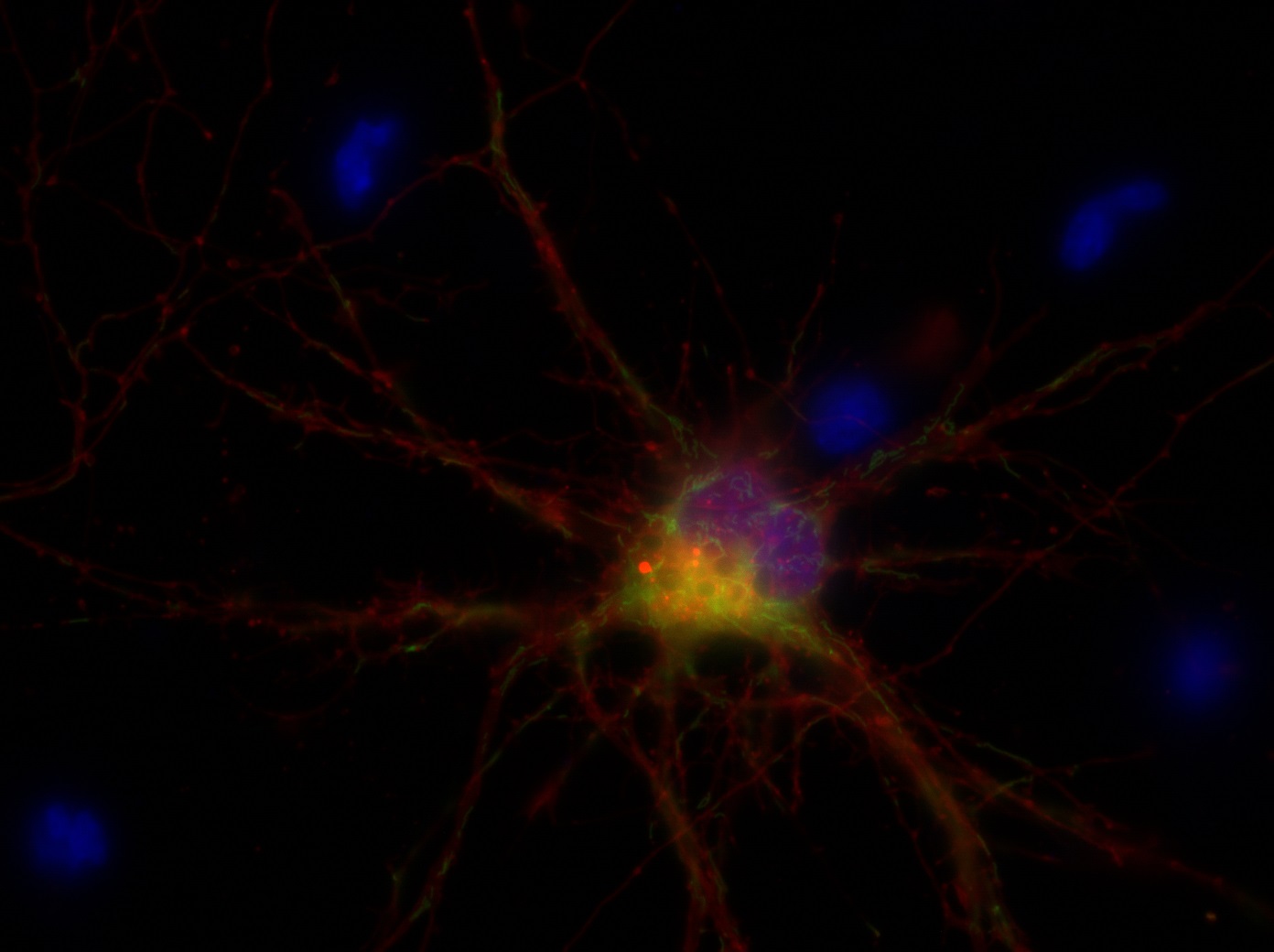
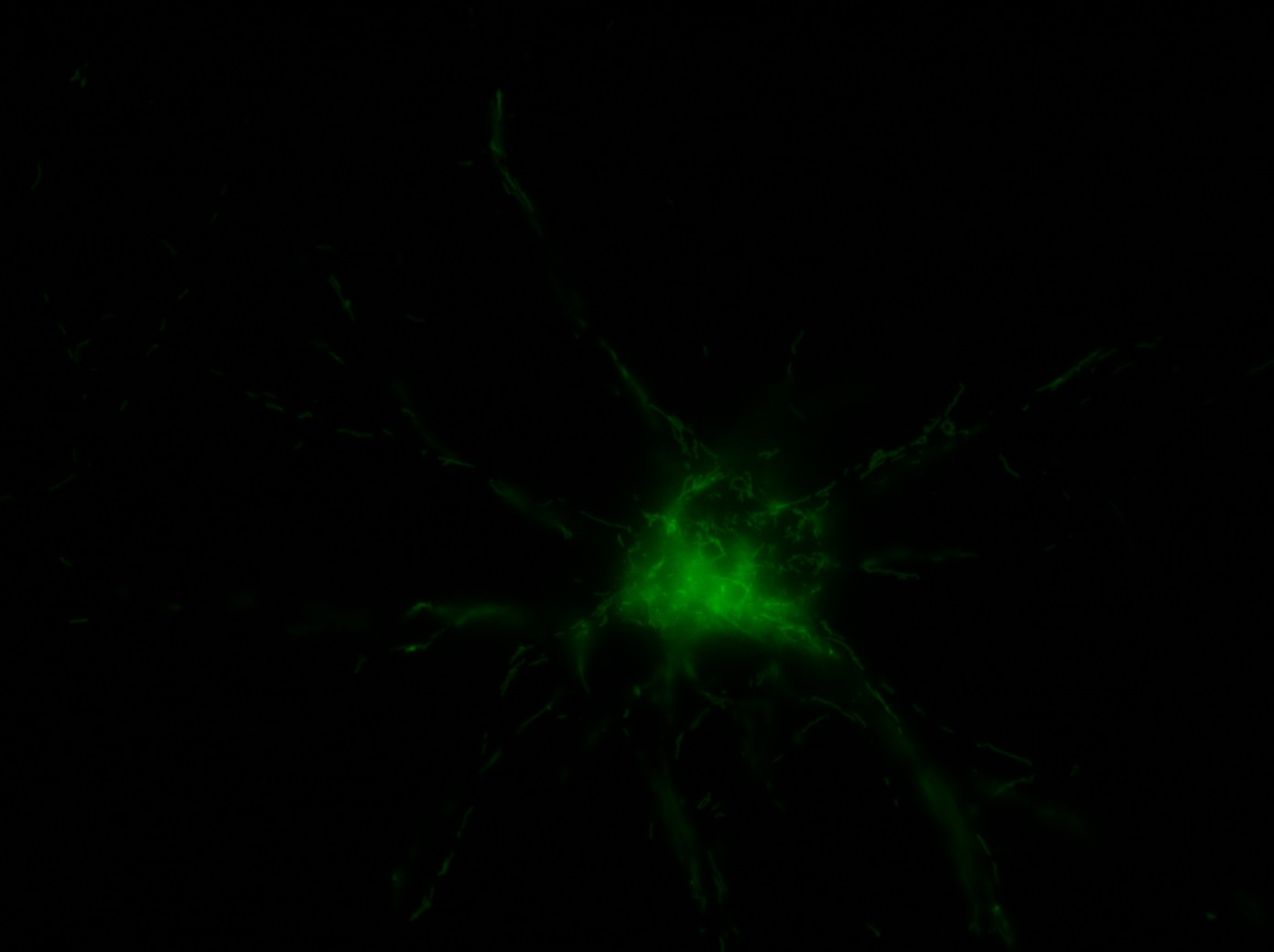
I was blinded toward each well and its corresponding transfection to eliminate potential bias in cell counting. The neurons were cultured from rat embryo hippocampus and we used three channels of the fluorescence microscope to inspect the neurons: the blue, green, and red channels. All the neurons were stained with Hoechst 33342, which only bound to DNA in the nucleus so that we could observe the nucleus in the blue channel. We were only interested in the transfected neurons, however, which all had GFP (green fluorescence protein) attached to Mff (mitochondria fission protein) so that we could see the mitochondria of transfected neurons in the green channel. Last but not least, the plasma membranes of transfected neurons contained Lck-mCherry so that we could see them in the red channel. Therefore, I used green channel to first locate the neurons that were transfected and then switched to blue and red channels to tell if that neuron was living or dead. Generally speaking, a round (elliptical) nucleus that was not uniform corresponded to a living cell and a condensed and uniform nucleus a dead cell. When looking at the red channel, I needed to first identify if the transfected cell was actually a neuron, which was what we were interested in. The wells might also contain glia cells, which could be identified in the red channel by having thicker and flatter processes than neurons. If the cell was indeed a neuron, broken dendrites in the red channel corresponded to dead neurons and intact dendrites living neurons. I posted some pictures of living and dead neurons in three different channels.


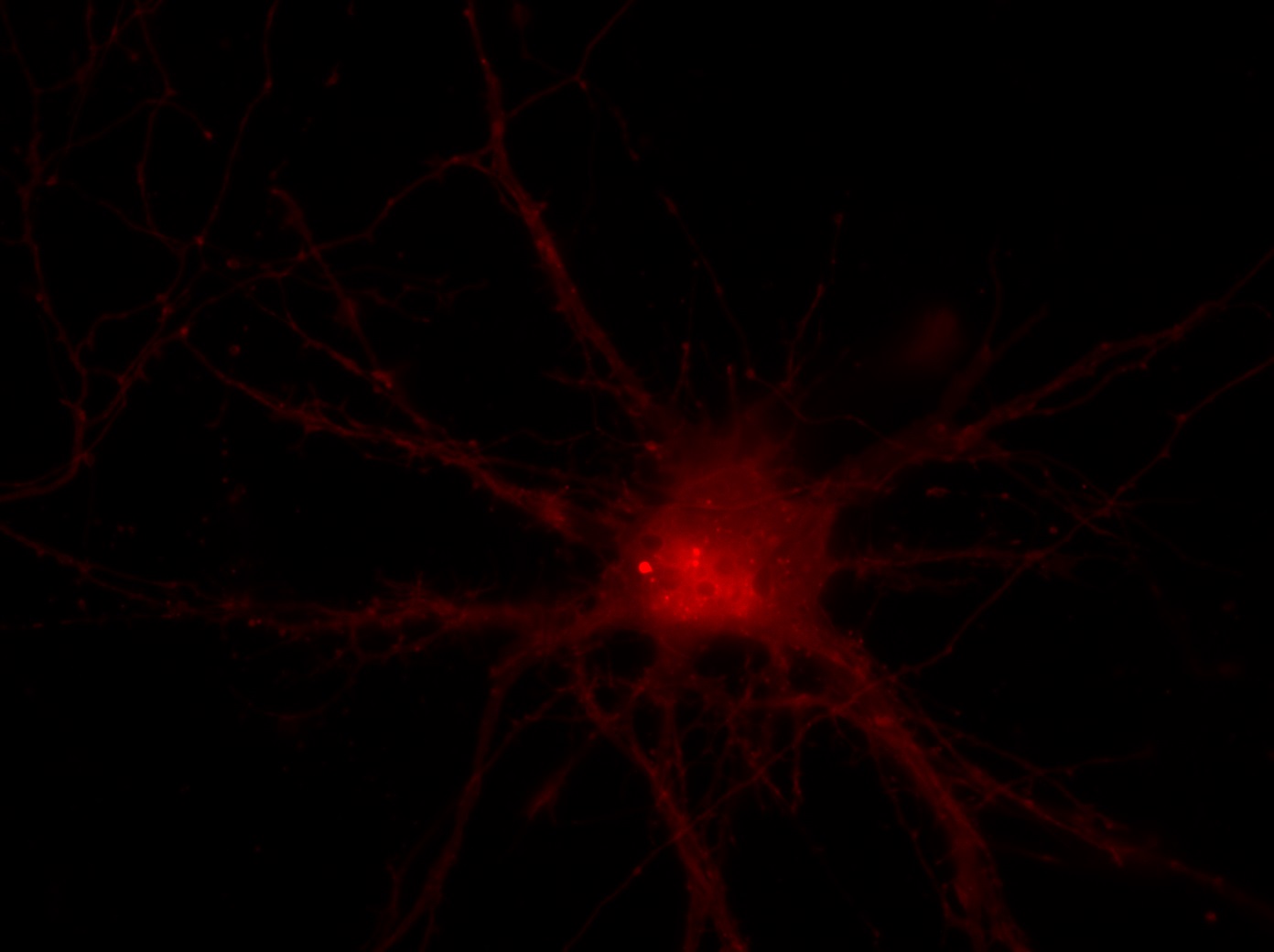
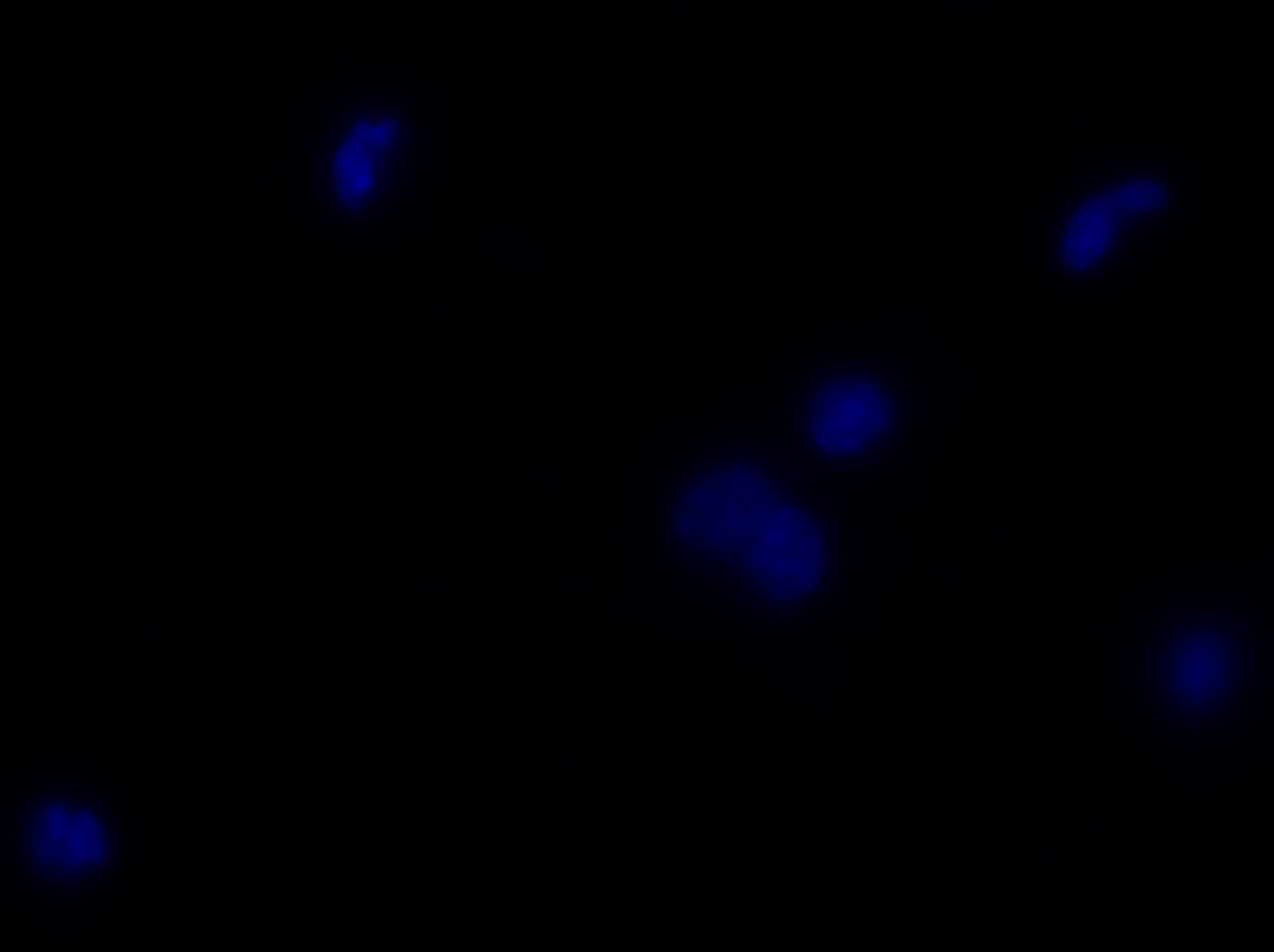
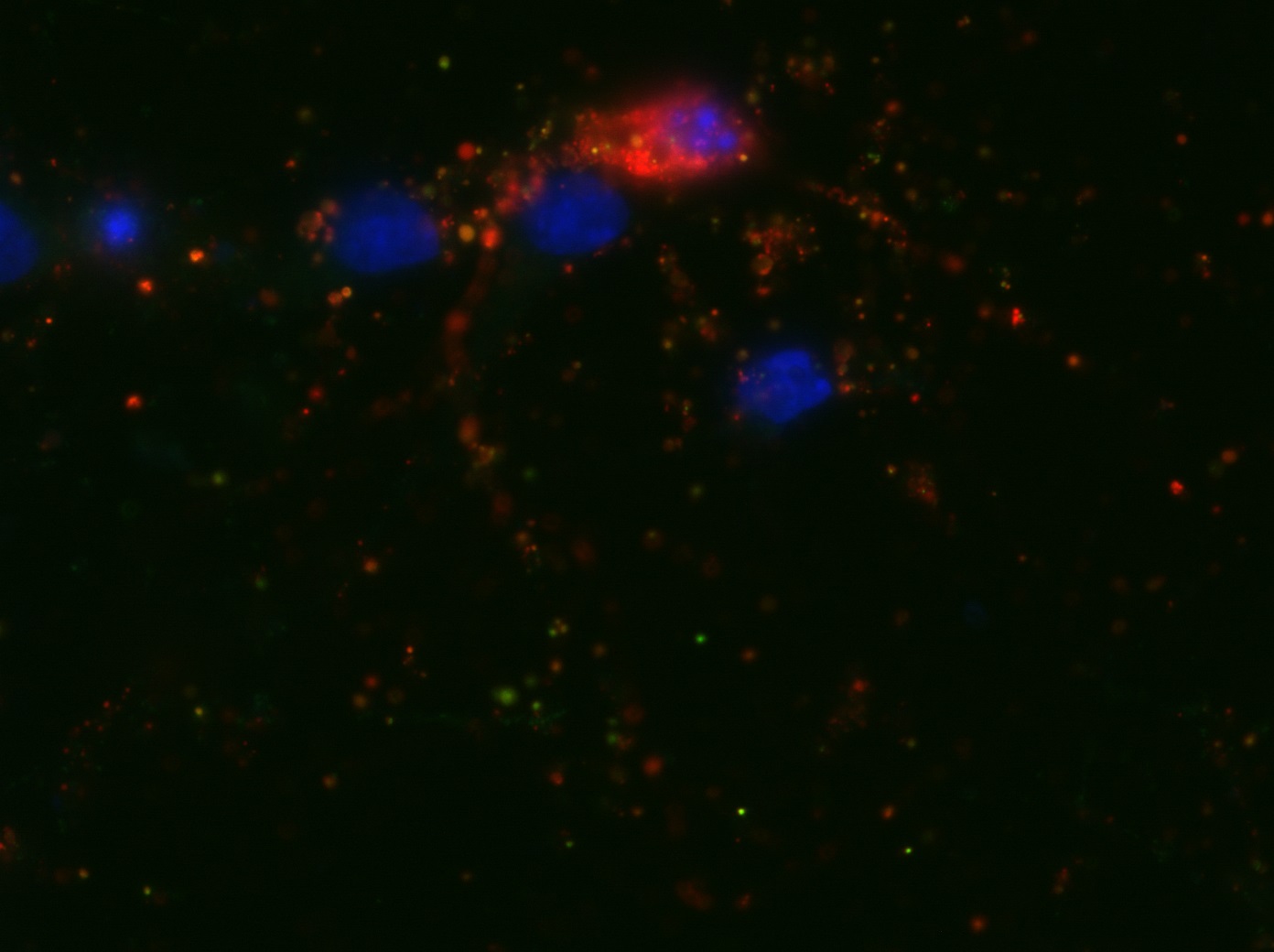
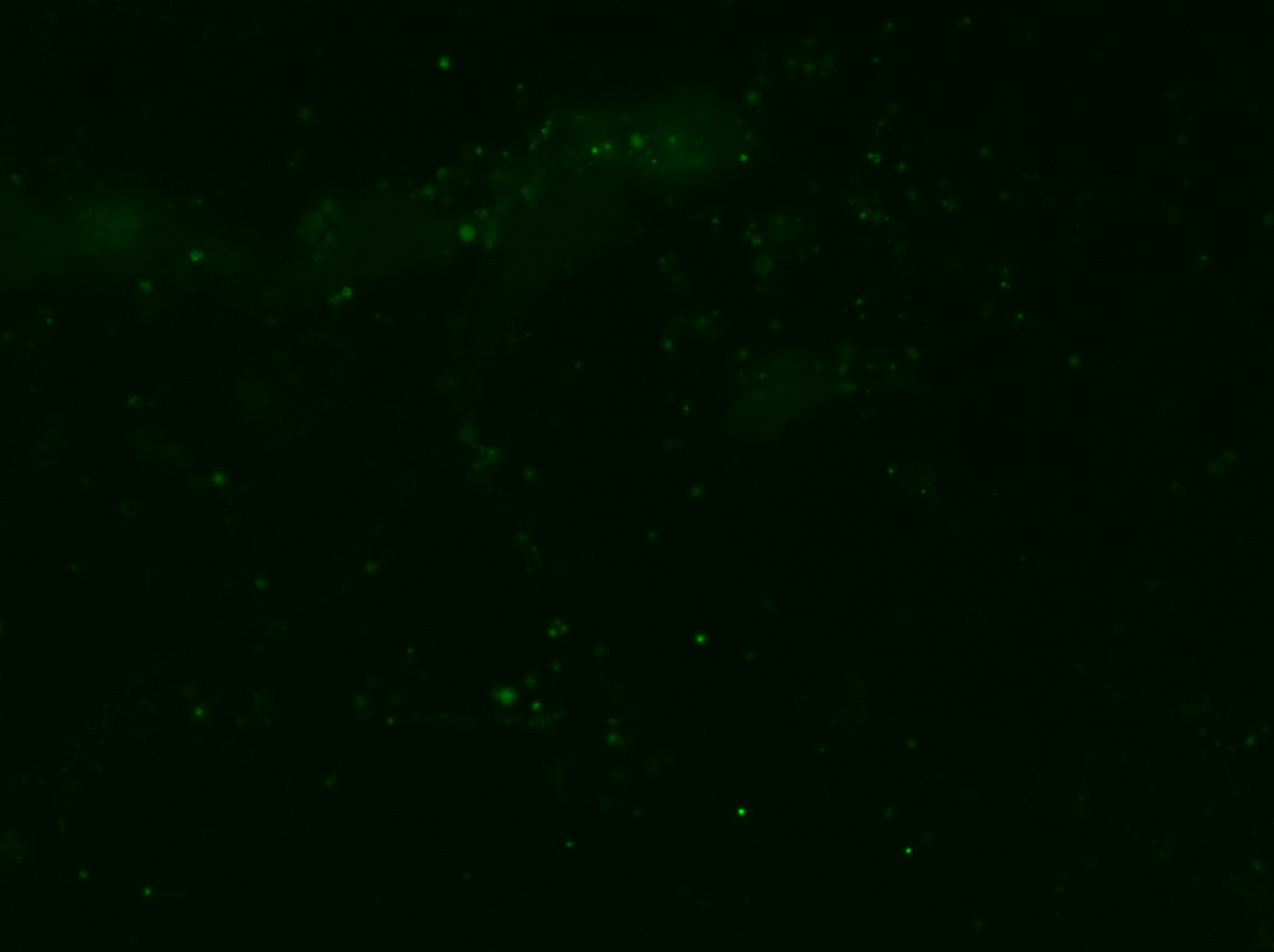
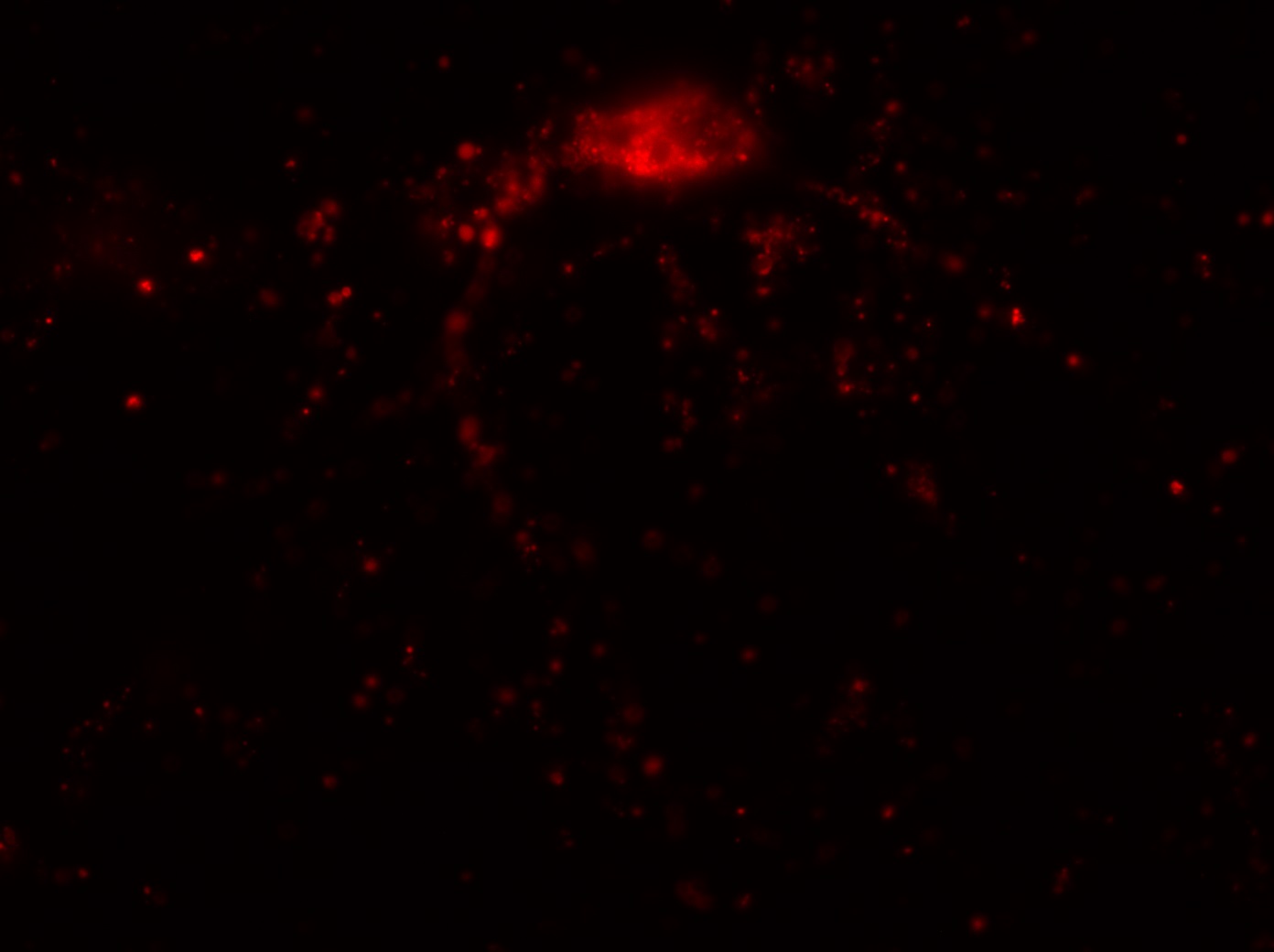
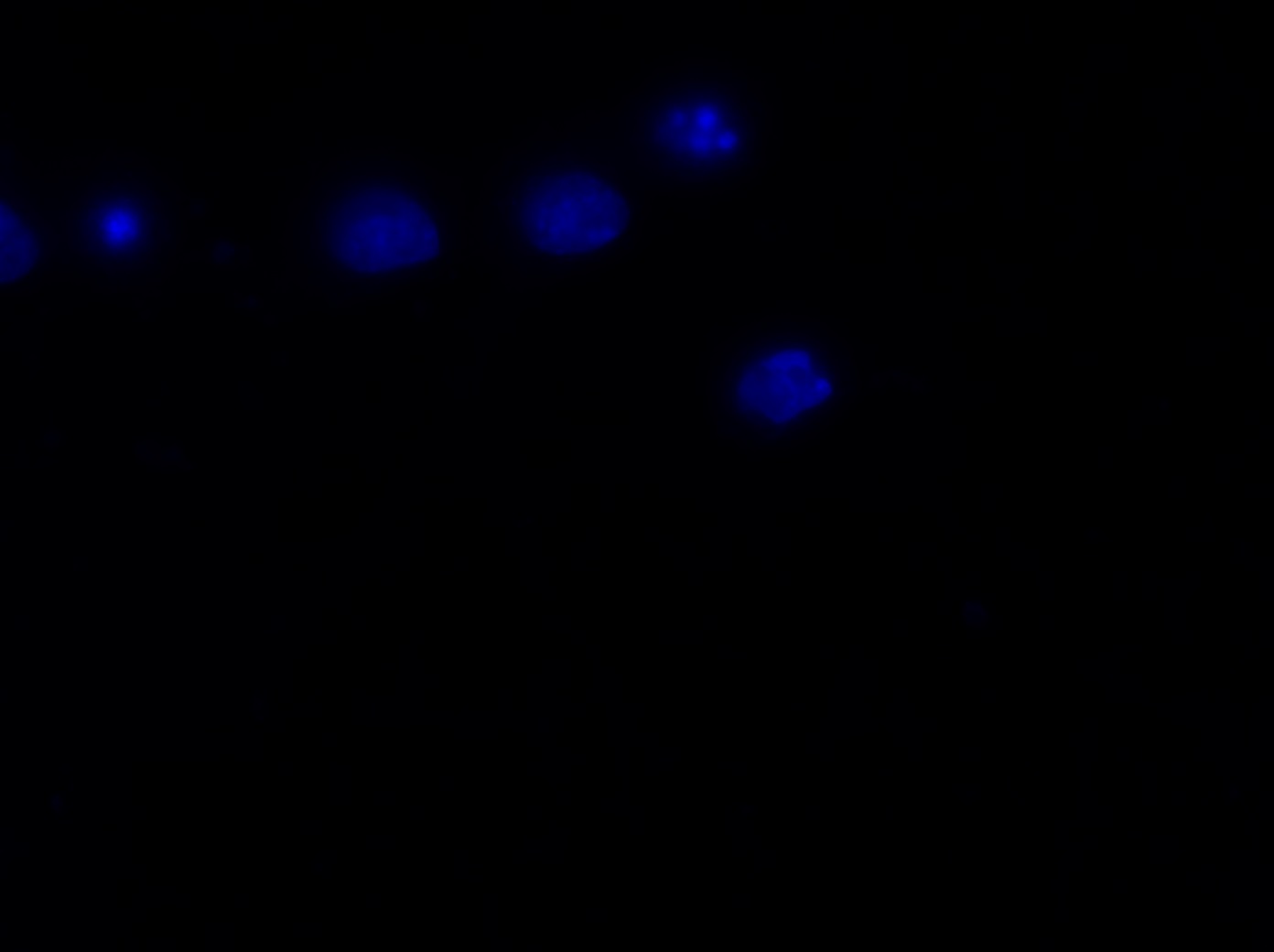

Major: Chemistry. Hometown:Centuria, Wisconsin.